
大田原综合征介绍
- 凤凰
-
 Topic Author
Topic Author
- Elite Member
-

- Posts: 232
大田原综合征介绍 was created by 凤凰
病因:部分可有脑结构异常(如脑穿通畸形、半侧巨脑征、Aicardi综合征、无脑回畸形、局灶性皮层发育不良等)、基因突变(如
STXBP1
(约占10-15%)、
SLC25A22
、
CDKL5
、
ARX
、
KCNQ2
、
SCN2A
、
GABRA1
、
CASK
、
KCNT1
、
SCN8A
、GABRB2、AARS、BRAT1、CACNA2D2、GNAO1、NECAP1、PIGA、PIGQ、SIK1等)或代谢异常(如
线粒体病
、非酮症性高甘氨酸血症、
吡哆醇(维生素B6)依赖性癫痫
、
磷酸吡哆醇(胺)氧化酶缺乏症
、肉碱棕榈酰转移酶缺乏等),三者之间可有交叉重叠[1-13]。
发病年龄:生后3个月内发病(多数在生后10天左右发病)。
发作特点:主要为强直痉挛发作(可不对称),部分可有局灶运动性发作,一般很少会有肌阵挛发作(注意需要和 早期肌阵挛脑病 加以鉴别),部分患儿后期会转变为 West 综合征 。
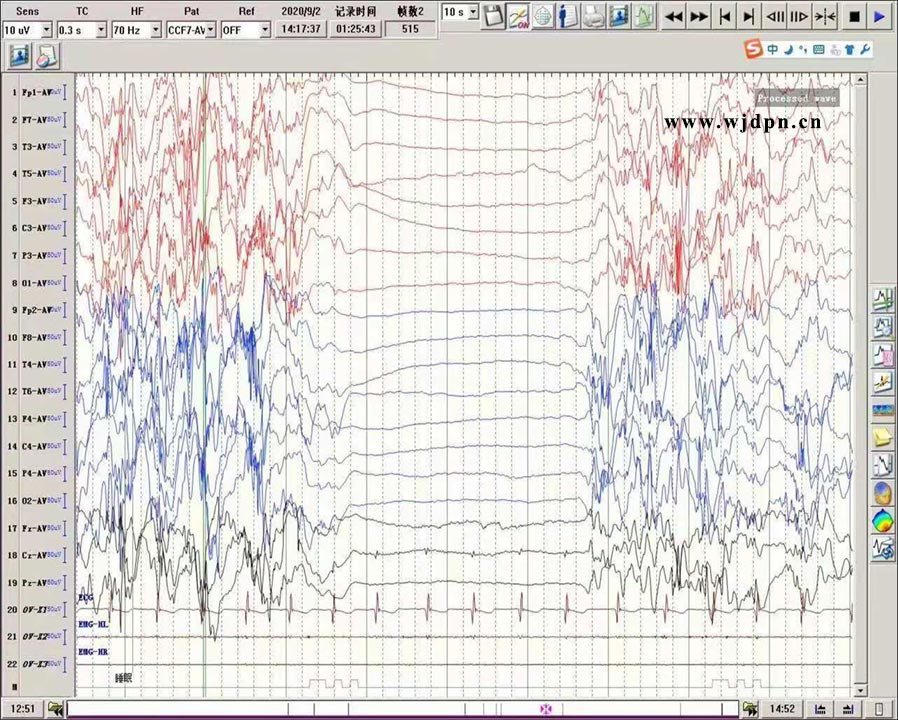
脑电图表现:发作间期:有 爆发抑制 (清醒和睡眠持续存在)。发作期:强直痉挛发作时可有高波幅慢波爆发,而后出现弥漫性低电压,如有局灶运动性发作时也可以有局灶性节律性放电 [14]。
头颅磁共振表现:部分可有脑结构发育异常,部分也可早期正常,后期出现萎缩等非特异性改变。
发育情况:常有严重发育落后。
注:考虑到 大田原综合征 和 早期肌阵挛脑病 在电临床方面具有较多的重叠,而且具有相似的潜在病因,对两者进一步区分不能再为临床决策或预后提供有价值的信息,2022年国际抗癫痫联盟(ILAE)已将 大田原综合征 和 早期肌阵挛脑病 都归类到早期婴儿发育性和癫痫性脑病(early infantile developmental and epileptic encephalopathy, EIDEE)中[15]。
早期婴儿发育性和癫痫性脑病诊断[15]:
必须具备的条件:强直和/或肌阵挛性癫痫发作;脑电图发作间期要有爆发抑制图形或多灶性放电,要有弥漫性慢波;出生3个月内起病(早产儿为纠正胎龄后);癫痫发作前或出现不久后就有发育障碍;整个病程中有神经发育异常,包括智力障碍。
需警惕可能不是的情况:起病前发育正常(当然回顾性评价有时较困难);神经系统检查正常(回顾性评价有时较困难,患儿频繁的发作以及抗癫痫药物治疗也会影响检查结果)。
参考文献
发病年龄:生后3个月内发病(多数在生后10天左右发病)。
发作特点:主要为强直痉挛发作(可不对称),部分可有局灶运动性发作,一般很少会有肌阵挛发作(注意需要和 早期肌阵挛脑病 加以鉴别),部分患儿后期会转变为 West 综合征 。
脑电图表现:发作间期:有 爆发抑制 (清醒和睡眠持续存在)。发作期:强直痉挛发作时可有高波幅慢波爆发,而后出现弥漫性低电压,如有局灶运动性发作时也可以有局灶性节律性放电 [14]。
{kind=link}
头颅磁共振表现:部分可有脑结构发育异常,部分也可早期正常,后期出现萎缩等非特异性改变。
发育情况:常有严重发育落后。
注:考虑到 大田原综合征 和 早期肌阵挛脑病 在电临床方面具有较多的重叠,而且具有相似的潜在病因,对两者进一步区分不能再为临床决策或预后提供有价值的信息,2022年国际抗癫痫联盟(ILAE)已将 大田原综合征 和 早期肌阵挛脑病 都归类到早期婴儿发育性和癫痫性脑病(early infantile developmental and epileptic encephalopathy, EIDEE)中[15]。
早期婴儿发育性和癫痫性脑病诊断[15]:
必须具备的条件:强直和/或肌阵挛性癫痫发作;脑电图发作间期要有爆发抑制图形或多灶性放电,要有弥漫性慢波;出生3个月内起病(早产儿为纠正胎龄后);癫痫发作前或出现不久后就有发育障碍;整个病程中有神经发育异常,包括智力障碍。
需警惕可能不是的情况:起病前发育正常(当然回顾性评价有时较困难);神经系统检查正常(回顾性评价有时较困难,患儿频繁的发作以及抗癫痫药物治疗也会影响检查结果)。
参考文献
- Stamberger, H., et al., STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy.Neurology, 2016. 86(10): p. 954-62.
- McTague, A., et al., The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol, 2016. 15(3): p. 304-16.
- Pavone, P., et al., Ohtahara syndrome with emphasis on recent genetic discovery.Brain Dev, 2012. 34(6): p. 459-68.
- Giordano, L., et al., Familial Ohtahara syndrome due to a novel ARX gene mutation.Am J Med Genet A, 2010.152A(12): p. 3133-7.
- Kato, M., et al., Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation.Epilepsia, 2013. 54(7): p. 1282-7.
- Touma, M., et al., Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings.Epilepsia, 2013. 54(5): p. e81-5.
- Kodera, H., et al., De novo GABRA1 mutations in Ohtahara and West syndromes.Epilepsia, 2016. 57(4): p. 566-73.
- Saitsu, H., et al., CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia, 2012. 53( 8 ): p. 1441-9.
- Yang, Y., et al., Phenotypic spectrum of patients with GABRB2 variants: from mild febrile seizures to severe epileptic encephalopathy. Dev Med Child Neurol, 2020. 62(10): p. 1213-1220.
- Saitsu, H., et al., Compound heterozygous BRAT1 mutations cause familial Ohtahara syndrome with hypertonia and microcephaly. J Hum Genet, 2014. 59(12): p. 687-90.
- Gertler, T., et al., KCNT1-Related Epilepsy, in GeneReviews((R)), M.P. Adam, et al., Editors. 1993: Seattle (WA).
- Martin, H.C., et al., Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet, 2014. 23(12): p. 3200-11.
- Alsahli, S., W. Al-Twaijri, and F. Al Mutairi, Confirming the pathogenicity of NECAP1 in early onset epileptic encephalopathy. Epilepsia Open, 2018. 3(4): p. 524-527.
- 李世绰. 临床诊疗指南, 癫痫病分册. 第2版. 北京 : 人民卫生出版社, 2015.
- Zuberi, S.M., et al., ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022.
25 May 2022 10:51
#1
Please Log in or Create an account to join the conversation.
Time to create page: 0.530 seconds