
 简体中文
简体中文  English
English Kearns-Sayre 综合征(KSS,Kearns-Sayre syndrome)
也有翻译为卡恩斯-塞尔综合征
病因:由于mtDNA(线粒体DNA)突变(主要是大片段缺失,个别也有点突变的报道),导致编码线粒体在氧化代谢过程中所必需的酶或载体发生障碍,属于线粒体脑肌病的一种亚型[1-2]。
临床表现:可多系统受累(眼、肌肉、神经系统、心脏、肾脏和内分泌腺体等能量需求较高的器官组织较容易累及),多在20岁之前发病,多为散发病例。当患儿同时具有眼外肌瘫痪、视网膜色素变性和心脏传导阻滞时,称为完全型Kearns-Sayre 综合征,当患者仅有眼外肌瘫痪或伴有其它一项时称为不完全型Kearns-Sayre 综合征。其它神经系统异常可包括认知功能障碍、卒中样发作、锥体外系症状(出现震颤、肌张力障碍)、神经性耳聋、共济失调以及延髓麻痹引起的吞咽困难等。不过此类患者较其它一些类型的线粒体脑肌病患者来说相对较少有癫痫发作[3-5]。
辅助检查[4, 6]:
1、实验室检查:血乳酸可增高(运动试验更具敏感性),此外脑脊液蛋白可增高(通常>1g/L)。
2、心电图:可表现为P-R间期延长、II或III度房室传导阻滞。
3、肌电图:肌电图多呈非特异性肌源性损害改变,神经传导速度一般正常。
4、肌肉活检:光镜下Gomori三色染色可见大量破碎样红纤维(ragged red fibe,RRF),细胞色素C氧化酶(COX)染色可见散在分布的COX缺失纤维。电镜下可见异常线粒体数目增多,线粒体嵴排列紊乱,有时线粒体内可见晶状格包涵体。
5、脑电图:有些可有非特性异常(可表现为散在慢波,有些也可出现癫痫样放电)。
6、头颅影像学:MRI可显示双侧皮层下白质、脑干、苍白球、丘脑、小脑等处长T2信号,另有些可有脑萎缩;MRS可见乳酸峰;少部分患者CT可显示基底节钙化。
MRI示例(经过临床、基因检测以及其它辅助检查相互验证):
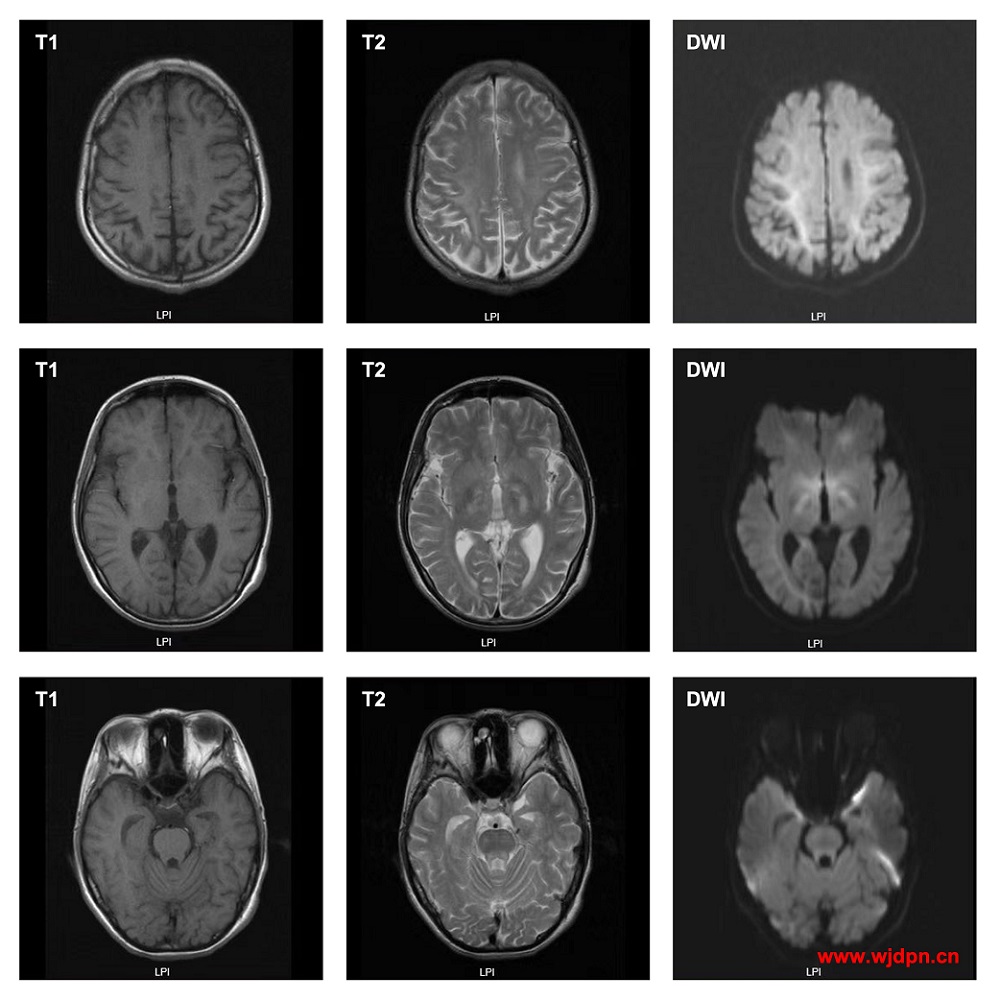
图示大脑白质、双侧丘脑、脑干等处可见多发斑片状等T1、略长T2以及DWI略高信号,边缘模糊,脑沟脑室略增宽。
7、其它:有些可有视力、听力等检查的异常。
参考文献
- Servidei, S., Mitochondrial encephalomyopathies: gene mutation. Neuromuscul Disord, 2001. 11(8): p. 774-9.
- Shoffner, J.M., Mitochondrial myopathy diagnosis. Neurol Clin, 2000. 18(1): p. 105-23.
- Finsterer, J., Central nervous system manifestations of mitochondrial disorders. Acta Neurol Scand, 2006. 114(4): p. 217-38.
- 包新华,姜玉武,张月华. 儿童神经病学. 第3版. 北京 : 人民卫生出版社, 2021.
- 刘晓燕. 临床脑电图学. 第2版. 北京 : 人民卫生出版社, 2017.
- 张旻玥, 李建萍, 苗玲. Kearns-Sayre综合征的临床表现、诊断与治疗[J]. 临床神经病学杂志, 2008(5):3.

